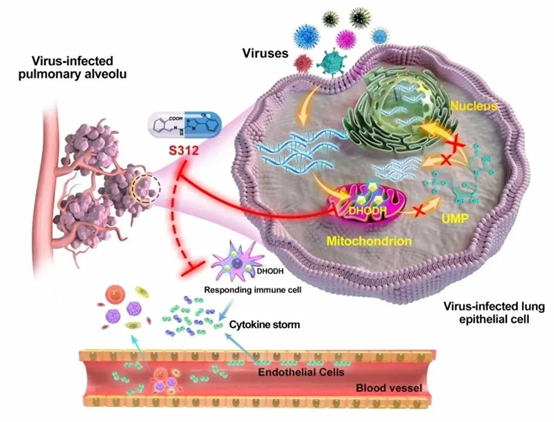
2020年3月13日,本团队和武汉大学病毒学国家重点实验室徐可团队合作,在生物预印本网站bioRxiv上发表题为Novel and potent inhibitors targeting DHODH, arate-limiting enzyme in de novo pyrimidine biosynthesis, are broad-spectrumantiviral against RNA viruses including newly emerged coronavirus SARS-CoV-2的未经同行评审文章,报道了一类的抗RNA病毒候选药物和老药品种。
2019年末至2020年初的美国流感病毒(Flu)和新型冠状病毒(SARS-CoV-2),2016年的寨卡病毒(Zika),2015年的中东呼吸综合征冠状病毒(MERS-CoV),2014年的埃博拉病毒(Ebola),2013年的流感病毒H7N9,2009年H1N1流感病毒,2003年的非典病毒(SARS-CoV)等RNA病毒,在全球范围内大规模爆发流行,给人们的生命健康带来严重危害的同时,也对国家及世界的经济发展带来巨大的冲击。截止2020年3月10日,新冠病毒已在全球范围内100多个国家感染113702人,并致4012人死亡,其中包括中国确诊病例80924人,死亡3140人1。SARS-CoV-2感染引起的肺炎(COVID-19)疫情已被WHO列为国际关注的突发公共卫生事件(PHEIC),且于3月11日被WHO正式宣布为世界大流行(Pandemic),但是目前仍没有研制出特效疫苗和抗病毒药物。病毒肆虐之下,寻找治疗新冠肺炎的高效、低毒的药物仍是目前我国乃至世界范围内对抗新冠病毒的当务之急。抗病毒药物可分为两大类:直接作用于病毒自身的抗病毒药物(Direct-acting antiviral agents, DAA)和靶向宿主因子的抗病毒药物(Host-targeting antiviral, HTA)。由于DAA药物具有病毒特异性,已有的DAA药物对新发病毒的治疗效果受限或完全无效,而重新研发新的DAA药物也是“远水解不了近渴”。而靶向宿主因子的HTA药物在控制新病毒疫情上就具有明显优势,病毒是寄生生物,必须依赖宿主复制,HTA药物不仅可有效抑制病毒核酸的快速复制,能对抗病毒耐药突变,做到“以不变应万变”。因此,具有广谱抗病毒药效的HTA药物的研发一直是抗感染新药研发领域追求的目标,但由于此类药物靶向宿主,同时也是双刃剑,需更多考虑安全性问题及较差的体外-体内药效转化问题,故此类药物的研发往往事倍功半,劳而少功2-6。
此外,流感和冠状病毒等RNA病毒的感染后期会诱发机体的过度免疫反应,因此,开展以宿主靶标如阻断核酸合成的一些关键酶、同时需兼顾调节自身免疫为药物研究靶标,进而获得可抑制病毒体内复制并同时可控制炎症因子风暴的药物,具有极大的临床需求和转化应用价值。
而针对流感和新型冠状病毒肺炎此类的急性病毒感染性疾病,需同时考虑短期内快速发现有效的临床治疗药物及中长期研发特效的候选新药作为新药储备。目前包括两种有效研究策略和方向:上市老药(有效药)和候选新药(特效药),前者可马上用于临床评价治病救人,后者则可作为长期研究和新药储备。基于此,两团队在靶向宿主的广谱抗病毒候选药物研究方面取得了一些进展:
1)发现靶向宿主的广谱抗病毒候选药物
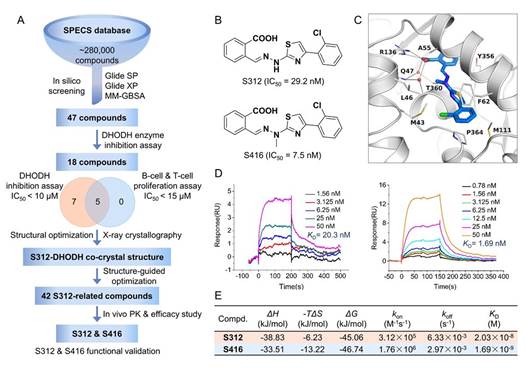
图1. 新型高效DHODH抑制剂的发现与设计

图2.DHODH抑制剂的广谱抗病毒活性
2)发现目前已报道的体外抑制活性最强的抗SARS-CoV-2候选化合物
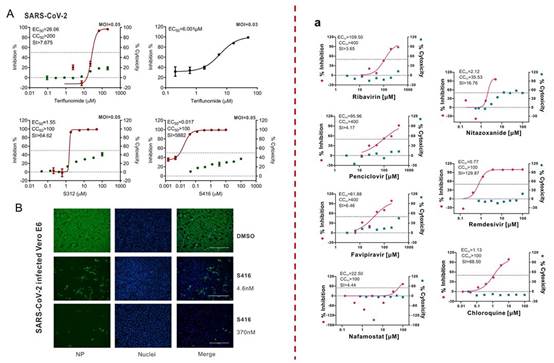
图3. DHODH抑制剂(左)与法匹拉韦, 瑞德西韦和氯喹等抑制剂(右)11的细胞水平抗SARS-CoV-2药效比较
同靶点的现有老药来氟米特的活性代谢产物特立氟胺(EC50=6.001μM,moi=0.03;26.06 μM,moi=0.05),在细胞水平抗SARS-CoV-2的EC50药效优于法匹拉韦(EC50=61.88 μM,moi=0.05)2倍以上,若通过首日负荷给药,以10mg/天的低剂量来氟米特12便可维持较高的血药治疗浓度,提示老药来氟米特或特立氟胺可能具有较好的抗新冠病毒的临床应用潜力。
相比核苷类药物的抗病毒多巧妙地采用“移花接木”策略(如核苷类似物样的假原料被掺入病毒基因组,导致病毒基因合成终止),本文设计的候选药物的抗病毒机制则是“釜底抽薪”,即阻断嘧啶碱基的从头合成过程,直接切断病毒RNA合成的原料供应。该候选药物前期已经完成初步毒理和药代试验,具有良好的成药性,获得国家“十三五”新药创制重大专项支持,作为新型抗流感候选药物正在开展临床前研究。
3)在动物体内证实DHODH抑制剂在致死性流感病毒感染小鼠中具有100%的保护效果
DHODH作为嘧啶从头合成途径的关键酶,是一潜在的广谱抗RNA病毒药物靶点。但其抑制剂在体内广谱抗病毒方面的应用可行性一直悬而未解。本文通过设计高活性的DHODH抑制剂,采用了从体内药效验证到DHODH基因敲除到病毒基因组复制量化检测,再到基因组复制所需不同原料补给的实验设计策略抽丝剥茧,最终在动物体内证实我们所设计的DHODH抑制剂是有效的RNA病毒广谱抑制剂(图4-5)。
分别使用甲型流感病毒感染的动物模型、达菲(Oseltamivir)耐药病毒株NAH275Y感染的动物模型及Oseltamivir不敏感病毒株SC09感染的动物模型证实了S312不仅具有与DAA药物同等的功效,并且在对抗DAA耐药病毒时更具优势;
使用CRISPR/Cas9基因敲除技术建立DHODH-/- A549细胞系,发现H1N1流感病毒在DHODH-/-细胞株上的增长较WT细胞株而言明显降低且不影响细胞的正常生长,证明DHODH是流感病毒复制所需的重要宿主酶;
使用流感病毒微型复制子系统来量化病毒基因组复制,发现S312和S416通过抑制病毒RNA复制发挥广谱抗病毒作用;
通过向流感病毒微型复制子系统分别添加A, G, U, C四种核苷酸观察其对流感病毒的拯救效果,发现只有添加U和C而非A和G可拯救流感病毒复制被抑制的效果,表明S312靶向嘧啶合成酶;通过分别向病毒复制子系统补充DHODH的底物二氢乳清酸(DHO)和产物乳清酸(ORO),发现ORO而非DHO的加入能剂量依赖性地拯救被抑制的流感病毒复制,表明S312通过靶向DHODH,抑制病毒基因组的复制。
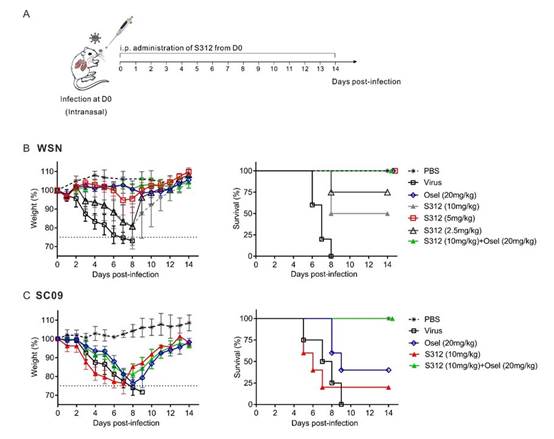
图4. S312在甲型流感病毒感染小鼠中的体内抗病毒活性
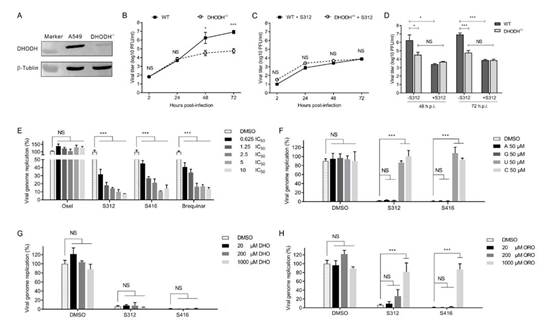
图5. 病毒基因组复制严重依赖于DHODH活性及从头合成嘧啶原料
4)调免疫、抗病毒“双管齐下”,对重症晚期疾病模型疗效优于DAA类药物
文章在流感病毒感染的重症小鼠体内模型上证明S312比达菲(Oseltamivir)更具显著优势的治疗效果(图6-7),进而提出DAA和HTA的组合用药是抗病毒治疗的一种有效策略。
在病毒感染中后期(感染后5-9天),达菲已失去治疗效果,而S312仍能发挥50%的小鼠拯救作用,若与达菲联合用药则能发挥100%的拯救效果。
急性病毒感染,如流感、新冠肺炎在感染晚期机体会产生过度炎症反应进而诱发“细胞因子风暴”。文章在病毒感染小鼠后第14天检测了达菲与S312联用前后的疾病小鼠肺泡灌洗液,发现DHODH抑制剂可降低IL6, MCP-1, IL5, KC/GRO(CXCL1), IL2, IFN-γ, IP-10, IL9, TNF-α, GM-CSF, EPO, IL12p70, MIP3α及IL17A/F等炎症因子的水平,预示DHODH抑制剂不仅可以有效切断核酸来源抑制病毒复制,还可能抑制过度炎症因子的表达和释放,在感染中晚期起到抗病毒抗炎双重效果。

图6. 与DAA药物Oseltamivir相比,S312在晚期和严重感染阶段更有效
本文不仅发现并验证了靶向宿主细胞的DHODH抑制剂能够通过抑制病毒复制和调节免疫两种途径发挥广谱抗病毒药效,还提示老药来氟米特或特立氟胺可能具有较好的抗新冠病毒的临床应用潜力,可满足目前新冠病毒肺炎临床急需。同时,新设计的候选药物S312和S416也有望开发成为抗RNA病毒的候选新药,为将来冠状病毒及其它RNA病毒的急性感染性疾病防控战做好了候选药物的储备。